Protein Based Phylogeny Trees
This web page was produced as an assignment for Genetics 677, an undergraduate course at University of Wisconsin: Madison.

Phylogeny.fr
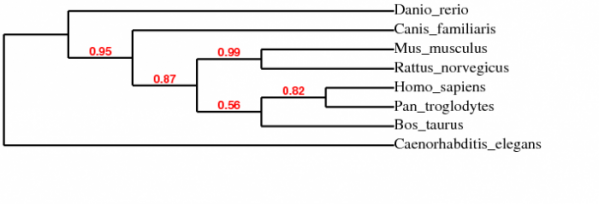
Figure 1 (Left 1,2,3,4,5,6). A phylogenetic tree of the protein homologs. The branchs are represented in a cladogram, which ignores the branch length. The red numbers are the branch support values (x100 to get percent).
Used: Phylogeny.fr: Robust Phylogenetic Analysis For The Non-Specialist to make phylogenetic trees using the one click program.
Analysis
The program Phylogeny.fr: Robust Phylogenetic Analysis For The Non-Specialist allows one to run a nice analysis for protein sequence homologs and DNA sequence homologs. However, the DNA sequence homologs are often times too long for the program. In addition, formatting can be often difficult to do, especially if you copy and paste your sequences into the program. In addition, it did not like the files I formatted and tried to upload even when I used the Fasta files. So, I have determined that it can be a touchy program especially when dealing with DNA sequences instead of protein sequences. I want to use the program to see if the phylogenetic tress would differ depending on the type of sequence used.
TreeTop
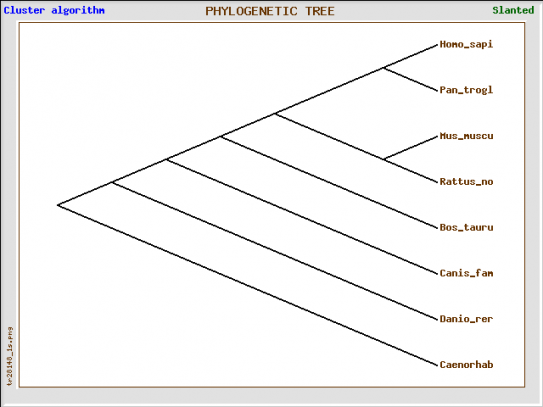
Figure 2. Phyolgenetic tree from TreeTop Phylogenetic TreePrediction.
Analysis
This is a really simple program and produces a simple result. One cannot edit the tree as well as with Phylogeny.fr: Robust Phylogenetic Analysis For The Non-Specialist, but it is a simple program that only takes the protein sequences input by the user, processes for 2 minutes, and outputs a quick and simple tree.
Pictures:
Dereeper A.*, Guignon V.*, Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M., Claverie J.M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W465-9. Epub 2008 Apr 19. 1
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, Mar 19;32(5):1792-7. 2
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, Apr;17(4):540-52. 3
Guindon S., Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, Oct;52(5):696-704. 4
Anisimova M., Gascuel O. Approximate likelihood ratio test for branchs: A fast, accurate and powerful alternative. Syst Biol. 2006, Aug;55(4):539-52. 5
Chevenet F., Brun C., Banuls AL., Jacq B., Chisten R. TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics. 2006, Oct 10;7:439. 6
TreeTop Phylogenetic TreePrediction. http://www.genebee.msu.su/services/phtree_reduced.html Figure 2
Megan Holler
[email protected]
Last Updated: 2/2/09
